Virusparies is an R package designed for visualizing outputs from VirusHunterGatherer, a data-driven virus discovery (DDVD) tool. It provides a set of plotting functions that aid in the interpretation and analysis of viral sequence data. The name draws inspiration from the hunter-gatherer metaphor, with “paries” derived from Latin meaning “wall”. It symbolizes the parietal art left by ancient hunters and gatherers on walls, summarizing their stories and beliefs.
VirusHunterGatherer is a computational pipeline designed for DDVD and is available on: https://github.com/lauberlab/VirusHunterGatherer. It involves two steps: (i) VirusHunter conducts sensitive homology-based detection of viral sequence reads in unprocessed data, identifying the most conserved regions of a virus, which serve as seeds for the (ii) Virusgatherer step that assembles full-length viral genome sequences.
To install the development version of Virusparies from GitHub, follow these steps:
### First install the "remotes" package
install.packages("remotes")
### Then install the Virusparies package
remotes::install_github("SergejRuff/Virusparies")! Bioconductor/CRAN version coming soon …
Virusparies includes the following functions:
ImportVirusTable(): Import VirusHunterGatherer
hittables into R.VhgBoxplot(): Boxplot plotting refSeq identity,
E-values or contig length for each group.VhgIdenFacetedScatterPlot(): Faceted scatter plot for
reference sequence identity vs -log10 of reference E-value.VhgIdentityScatterPlot(): Scatter Plot for reference
Identity vs -log10 of reference E-value.VhgRunsBarplot(): Bar plot showing how many unique Runs
map against each virus.VhgSumHitsBarplot(): Bar plot for the sum of hits for
each virus found in group.VgConLenViolin(): Violin plot to visualize the
distribution of contig lengths.VhgRunsTable() Generate a GT-table for
VhRunsBarplot.VhgTabularRasa() Generate custome GT-tables.The table functions generate GT objects, which can be further manipulated using the GT package (see the Details section for more information).
ExportVirusDataFrame(): Export data frames.ExportVirusGt(): Export graphical tables.ExportVirusPlot(): Export plots.CombineHittables(): Combine hittables.Current_ICTV(): Verify the version of ICTV data
currently in use.New_ICTV(): Assign Custom ICTV Data for Use in
Virusparies.SummarizeViralStats(): Generate summary stats outside
of plot functions.VhgAddPhylum(): Extract phylum information.VhgGetSubject(): Process and Count Viral Subjects
within Groups.VhgPreprocessTaxa(): Process ViralRefSeq_taxonomy
column.VhgSubsetHittable(): Filter VirusHunterGatherer data
based on user’s own criteria.The Virusparies package provides a set of plotting functions tailored for visualizing VirusHunterGatherer hittables and generating graphical tables (GT) for results generated by Virusparies or the user. It includes functions for importing VirusHunterGatherer hittables and exporting both plots and graphical tables. This section explains the functionality of each function in detail.
Each function accepts a VirusHunterGatherer file (vh_file argument), a column for grouping on the x-axis (x_column or groupby argument), a y_column argument and a cutoff for filtering observations below the user-defined E-value threshold. Key points to note: - Accepted x_column (or groupby) arguments are “best_query” (only for VirusHunter hittables) or “ViralRefSeq_taxonomy” (Default: “best_query”). - The cutoff is defined by the “ViralRefSeq_E” column. - The default cutoff E-value is 1e⁻⁵, but users can set their own cutoff E-values. - Cutoffs are used for filtering observations above the defined threshold. Unless the thing being used for plotting is the “ViralRefSeq_E” column (see :Boxplot 1: “ViralRefSeq_E” or both scatterplots as an example). In those cases the unfiltered hittable is plotted and the cutoff is used to highlight the proportion of E-values above and below the threshold. - Each plot includes a legend based on the Phylum of each virus group (expect for the faceted scatter plot, where each point is colored based on whether the observation is above or below the threshold). - Both table and plot functions provide arguments for customization.
Table functions return a GT object, and plot functions returns a ggplot2 object for further manipulation. Some plots also generate and return summary statistics or filtered VirusHunterGatherer files for further downstream analysis.
First, we load the package into R:
### load Virusparies into R
library(Virusparies)VirusHunterGatherer hittables are tab-separated values (TSV) files
that can be imported with the ImportVirusTable() function.
Here, we use the example VirusHunter and VirusGatherer hittable files
included in the Virusparies package:
### Import VirusHunter Hittable.
path <- system.file("extdata", "virushunter.tsv", package = "Virusparies")
vh_file <- ImportVirusTable(path)
print(head(vh_file)) # print head of hunter files
#> SRA_run SRA_sample SRA_study host_taxon host_taxid num_queries num_hits best_E
#> 1 SRR10822543 SRS5937532 SRP239389 Homo_sapiens 9606 8 12 4.5e-05
#> 2 SRR12567985 SRS7305728 SRP279687 Homo_sapiens 9606 6 9 9.7e-13
#> 3 SRR12567985 SRS7305728 SRP279687 Homo_sapiens 9606 6 5 1.4e-08
#> 4 SRR10822560 SRS5937549 SRP239389 Homo_sapiens 9606 11 3 1.1e-08
#> 5 SRR12567985 SRS7305728 SRP279687 Homo_sapiens 9606 6 8 3.2e-10
#> 6 SRR10822546 SRS5937535 SRP239389 Homo_sapiens 9606 7 20 1.0e-01
#> best_query ViralRefSeq_E ViralRefSeq_ident ViralRefSeq_aLen.sLen ViralRefSeq_contigs
#> 1 Anello_ORF1core 2.59e-38 93.8 192 / 194 1
#> 2 Anello_ORF1core 8.44e-36 77.3 225 / 227 6
#> 3 Anello_ORF1core 3.95e-34 70.1 231 / 232 1
#> 4 Anello_ORF1core 3.69e-31 68.6 210 / 216 1
#> 5 Anello_ORF1core 5.85e-31 76.9 195 / 201 3
#> 6 Anello_ORF1core 8.63e-31 79.4 189 / 237 1
#> ViralRefSeq_subject
#> 1 gi:1464307144|Torque teno mini virus 11 isolate LIL-y2 ORF2, ORF1, and ORF3 genes, complete cds
#> 2 gi:1464307216|Torque teno midi virus 11 DNA, complete genome, isolate: MDJN47
#> 3 gi:1464307144|Torque teno mini virus 11 isolate LIL-y2 ORF2, ORF1, and ORF3 genes, complete cds
#> 4 gi:134133206|Torque teno midi virus 1, complete genome
#> 5 gi:1464307186|Torque teno midi virus 5 DNA, complete genome, isolate: MDJHem2
#> 6 gi:1464307221|Torque teno midi virus 12 DNA, complete genome, isolate: MDJN51
#> ViralRefSeq_taxonomy date_analyzed
#> 1 taxid:2065037|Betatorquevirus|Anelloviridae 2022-05-17
#> 2 taxid:2065052|Gammatorquevirus|Anelloviridae 2022-05-06
#> 3 taxid:2065037|Betatorquevirus|Anelloviridae 2022-05-06
#> 4 taxid:687379|Gammatorquevirus|Anelloviridae 2022-05-17
#> 5 taxid:2065046|Gammatorquevirus|Anelloviridae 2022-05-06
#> 6 taxid:2065053|Gammatorquevirus|Anelloviridae 2022-05-17
### Import VirusGatherer Hittable.
path2 <- system.file("extdata", "virusgatherer.tsv", package = "Virusparies")
vg_file <- ImportVirusTable(path2)
print(head(vg_file)) # print head of gatherer files
#> SRA_run SRA_sample SRA_study host_taxon host_taxid contig_id
#> 1 ERR206007 ERS074208 ERP000373 Homo sapiens 9606 ERR206007_cap3_Contig-1
#> 2 ERR206007 ERS074208 ERP000373 Homo sapiens 9606 ERR206007_cap3_Contig-2
#> 3 ERR206007 ERS074208 ERP000373 Homo sapiens 9606 ERR206007_cap3_Contig-3
#> 4 ERR206007 ERS074208 ERP000373 Homo sapiens 9606 ERR206007_cap3_Contig-4
#> 5 ERR206021 ERS074222 ERP000373 Homo sapiens 9606 ERR206021_cap3_Contig-1
#> 6 SRR10822543 SRS5937532 SRP239389 Homo sapiens 9606 SRR10822543_cap3_Contig-1
#> contig_len ViralRefSeq_E ViralRefSeq_ident ViralRefSeq_aLen
#> 1 603 4.22e-65 92.453 106
#> 2 461 3.22e-65 85.612 139
#> 3 364 2.46e-53 76.531 98
#> 4 334 9.87e-67 93.636 110
#> 5 323 8.91e-45 65.421 107
#> 6 3321 0.00e+00 86.058 789
#> ViralRefSeq_subject
#> 1 acc:YP_009505712|Orf1 [Torque teno virus 5]
#> 2 acc:YP_003587853|hypothetical protein TTV10_gp4 [Torque teno virus 10]
#> 3 acc:YP_003587868|ORF1 [Torque teno virus 3]
#> 4 acc:YP_009505715|Orf1 [Torque teno virus 11]
#> 5 acc:YP_009505729|unnamed protein product [Torque teno virus 24]
#> 6 acc:YP_009173866|polymerase [Hepatitis B virus]
#> ViralRefSeq_taxonomy
#> 1 taxid:687344|Alphatorquevirus homin5|Alphatorquevirus|Anelloviridae
#> 2 taxid:687349|Alphatorquevirus homin10|Alphatorquevirus|Anelloviridae
#> 3 taxid:687342|Alphatorquevirus homin3|Alphatorquevirus|Anelloviridae
#> 4 taxid:687350|Alphatorquevirus|Anelloviridae
#> 5 taxid:687363|Alphatorquevirus homin24|Alphatorquevirus|Anelloviridae
#> 6 taxid:10407|Orthohepadnavirus|Hepadnaviridae|Blubervirales|Revtraviricetes|Artverviricota|Pararnavirae|Riboviria
#> date_analyzed
#> 1 2024-05-18
#> 2 2024-05-18
#> 3 2024-05-18
#> 4 2024-05-18
#> 5 2024-05-17
#> 6 2024-05-18
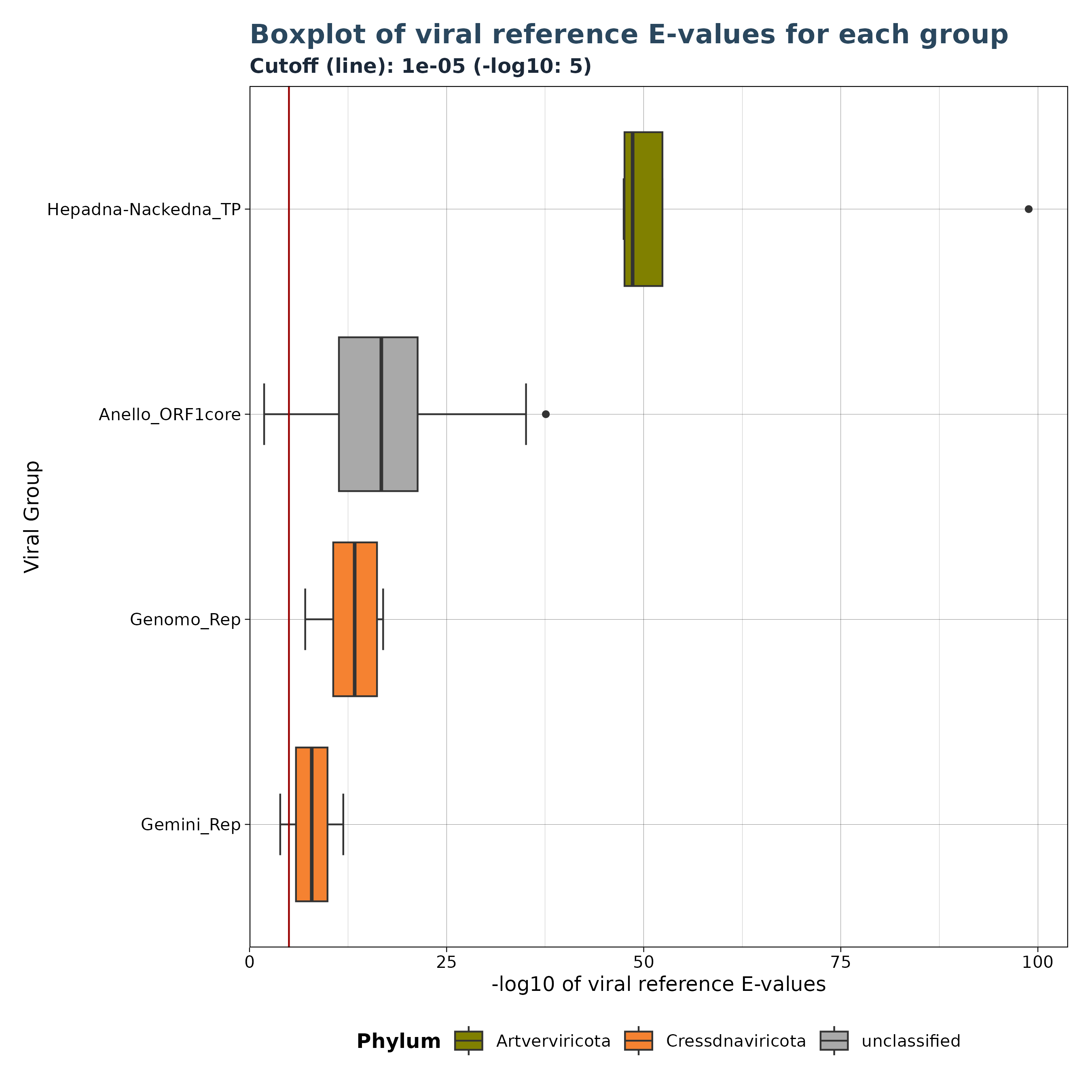
The VhgBoxplot() function generates three versions of a
boxplot depending on the provided y_column
argument.
Accepted y_column arguments are: - “ViralRefSeq_E”: Distribution of viral reference E-values. - “ViralRefSeq_ident”: Distribution of sequence identity to the closest viral reference. - “contig_len” (Gatherer Tables only): Distribution of contig lengths.
Below are 4 examples for different boxplots.
### Plot 1 for evalues
plot1 <- VhgBoxplot(vh_file, x_column = "best_query", y_column = "ViralRefSeq_E")
plot1
### Plot 2 for identity
plot2 <- VhgBoxplot(vh_file, x_column = "best_query", y_column = "ViralRefSeq_ident")
plot2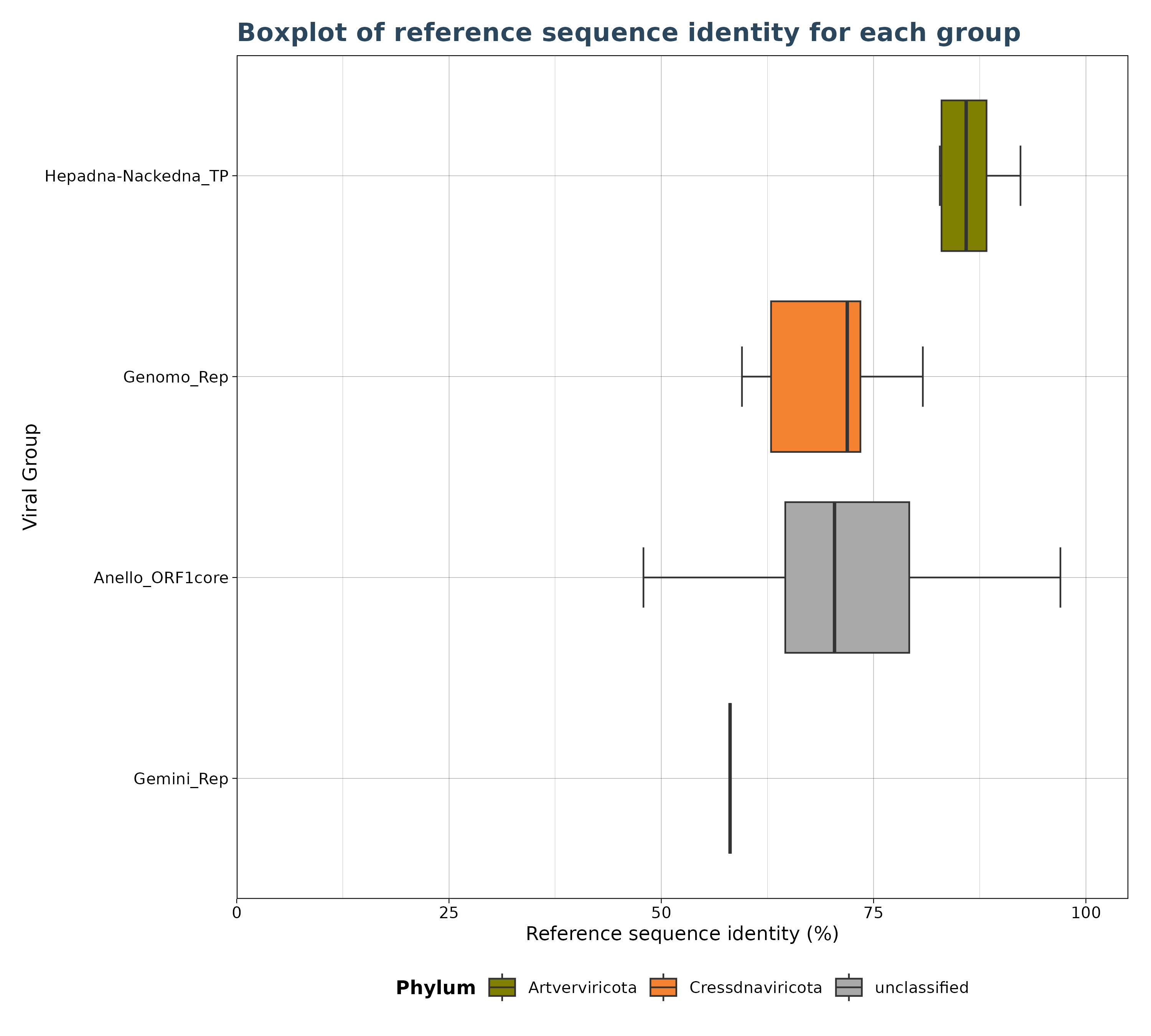
Plots and tables in Virusparies are highly customizable. This is true for all functions in Virusparies. Example 3 demonstrates it by changing the text of the subtitle and axis-labels, changing the position of the legend and changing the background theme.
### Plot 3 custom arguments used
plot3 <- VhgBoxplot(vh_file,
x_column = "best_query",
y_column = "ViralRefSeq_E",
theme_choice = "grey",
subtitle = "Custom subtitle: Identity for custom query",
xlabel = "Custom x-axis label: Custom query",
ylabel = "Custom y-axis label: Viral Reference Evalue in -log10 scale",
legend_position = "right")
plot3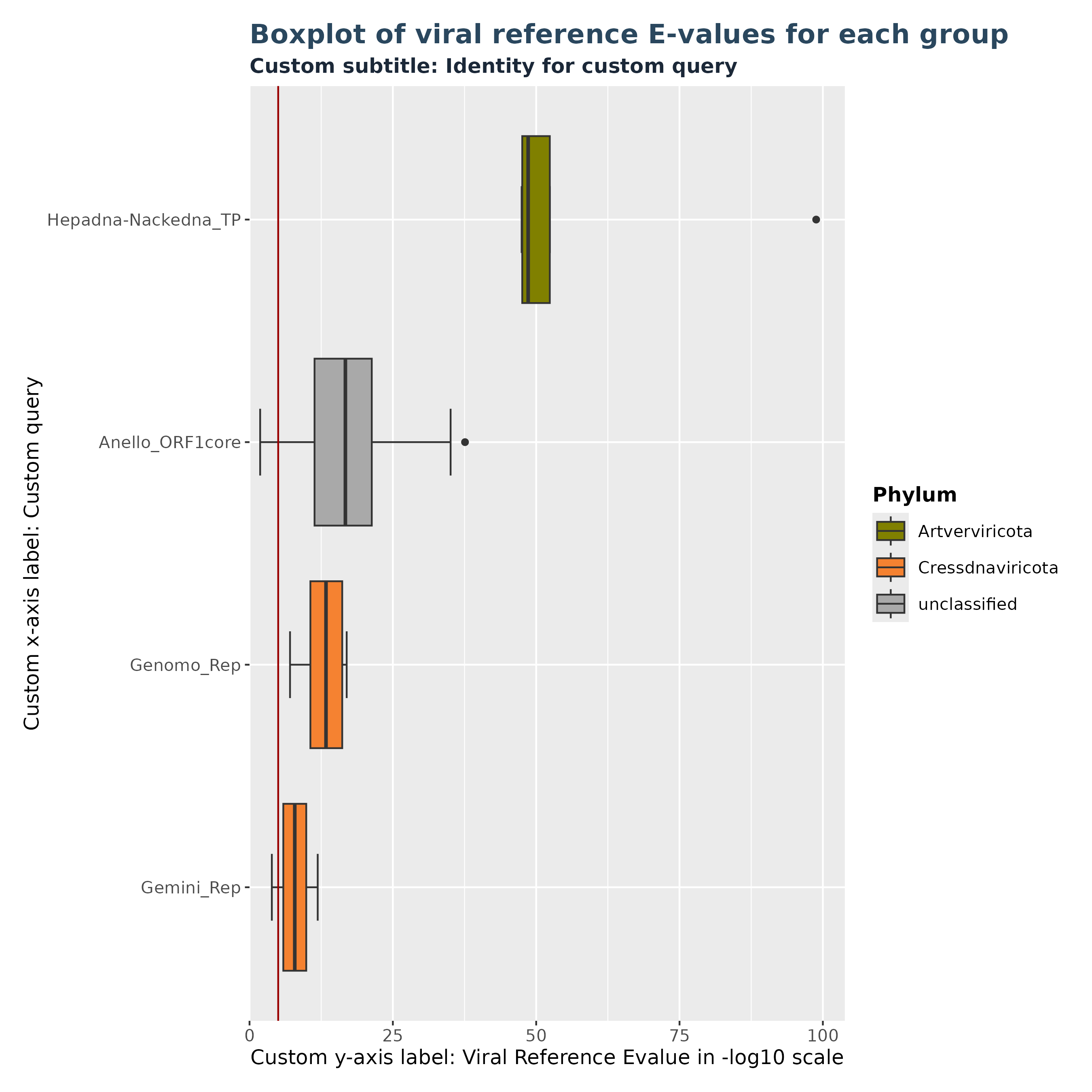
### Plot 5: Virusgatherer plot for SRA_runs agains contig length
plot4 <- VhgBoxplot(vg_file,x_column = "ViralRefSeq_taxonomy",y_column = "contig_len")
plot4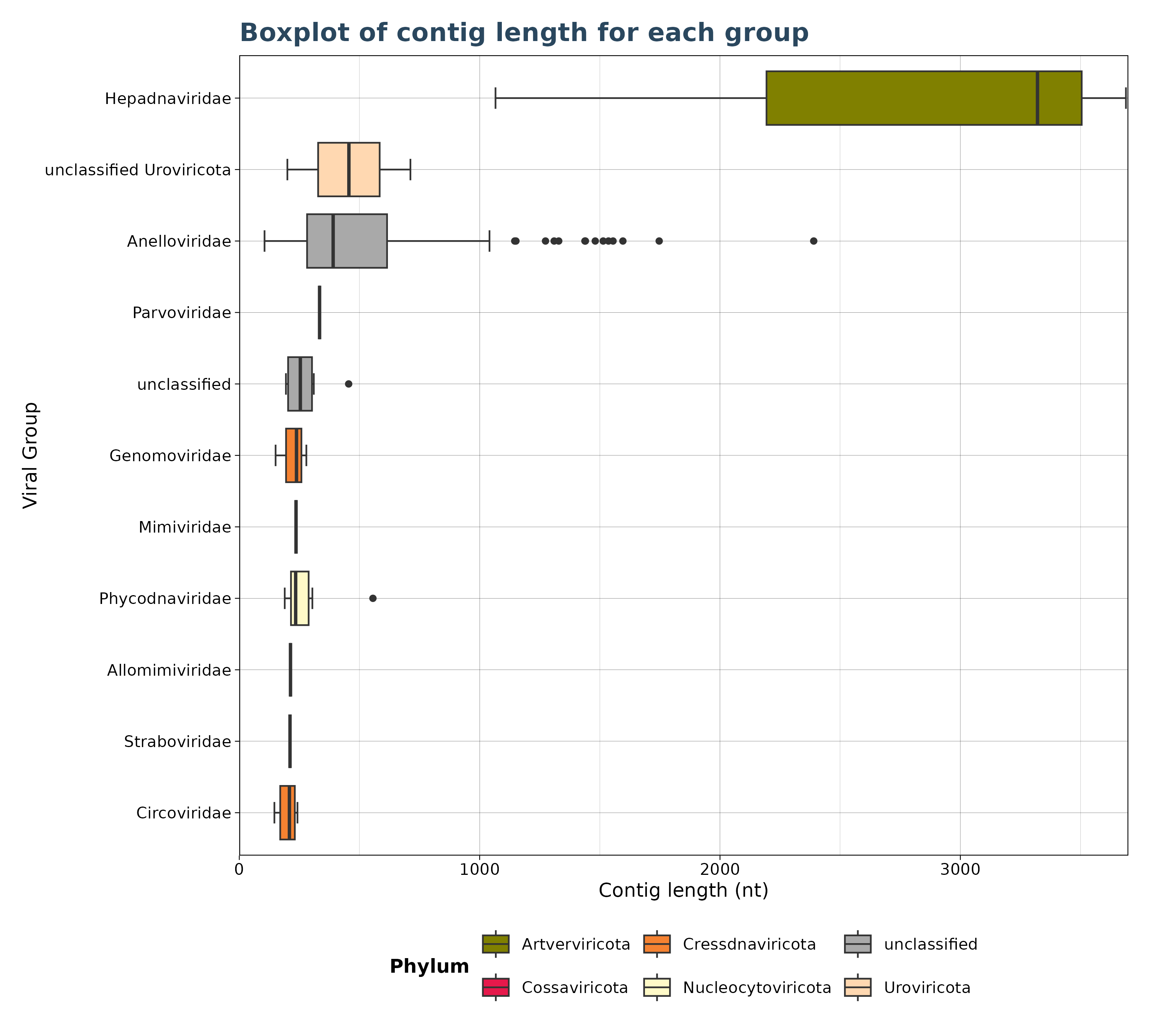
VhgIdenFacetedScatterPlot() generates a scatter plot
with viral reference identity (“ViralRefSeq_ident” column) on the x-axis
and the -log10 of Viral reference E-values (“ViralRefSeq_E”) on the
y-axis. Observations are colored based on whether they are below or
above the specified threshold. By default blue points indicate
observations with E-values below the threshold and red indicates points
above the threshold. The VhgIdenFacetedScatterPlot()
creates faceted plots for each group defined by
groupby. This allows the user to better
separate virus groups into different plots,especially in cases where
multiple groups cluster to closely together and are no longer
distinguishable in the VhgIdentityScatterPlot() plot.
### Generate Plot
plot <- VhgIdenFacetedScatterPlot(vh_file,cutoff = 1e-5)
plot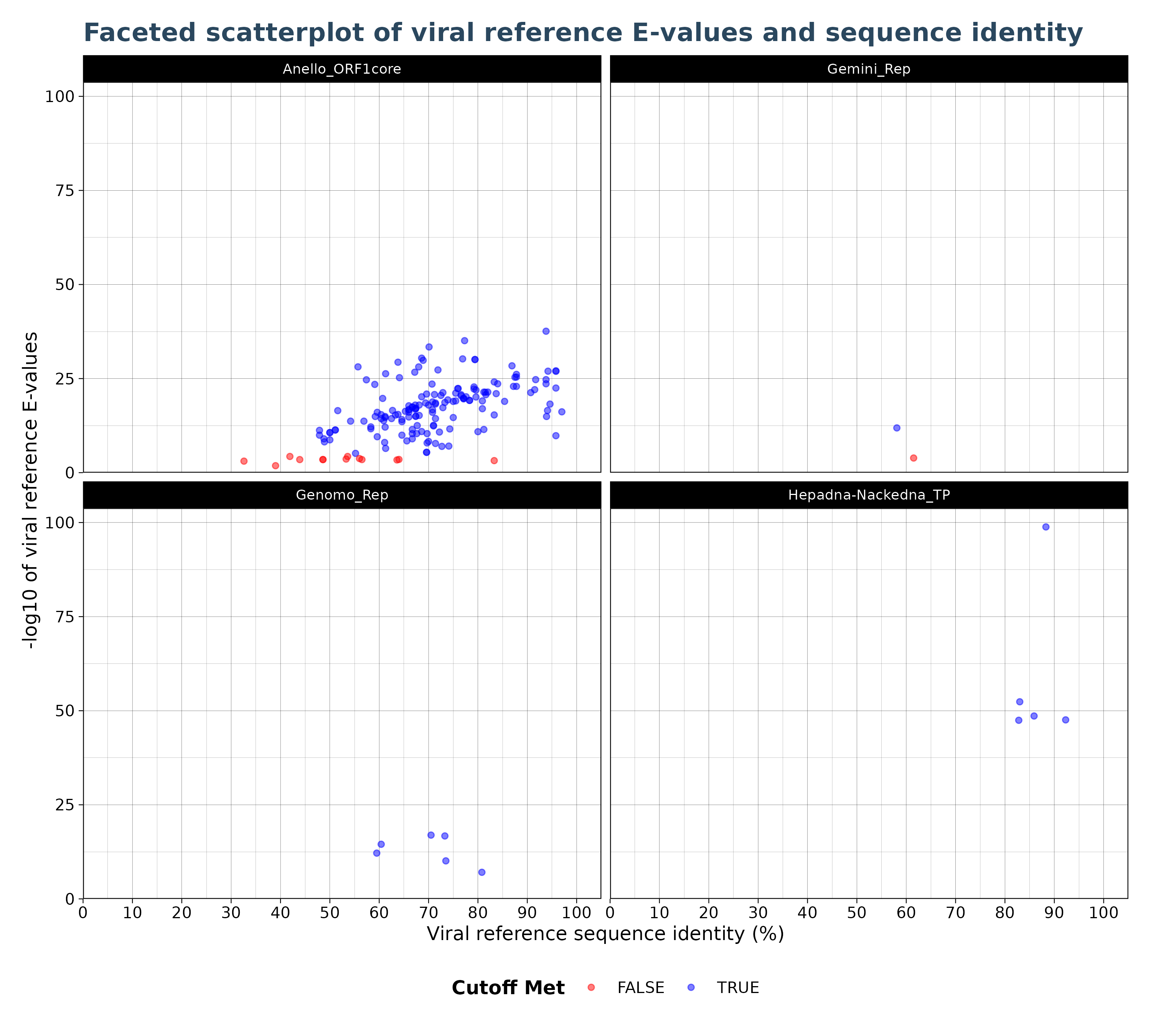
VhgIdentityScatterPlot() generates a scatter plot with
viral reference sequence identity (“ViralRefSeq_ident” column) on the
x-axis and the -log10 of viral reference E-values (“ViralRefSeq_E”) on
the y-axis.
A red horizontal line (see figure below) indicates whether an observation is above or below threshold.
scate_plot <- VhgIdentityScatterPlot(vh_file,cutoff = 1e-5,
highlight_groups = c("Gemini_Rep","Genomo_Rep"))
plot(scate_plot$plot)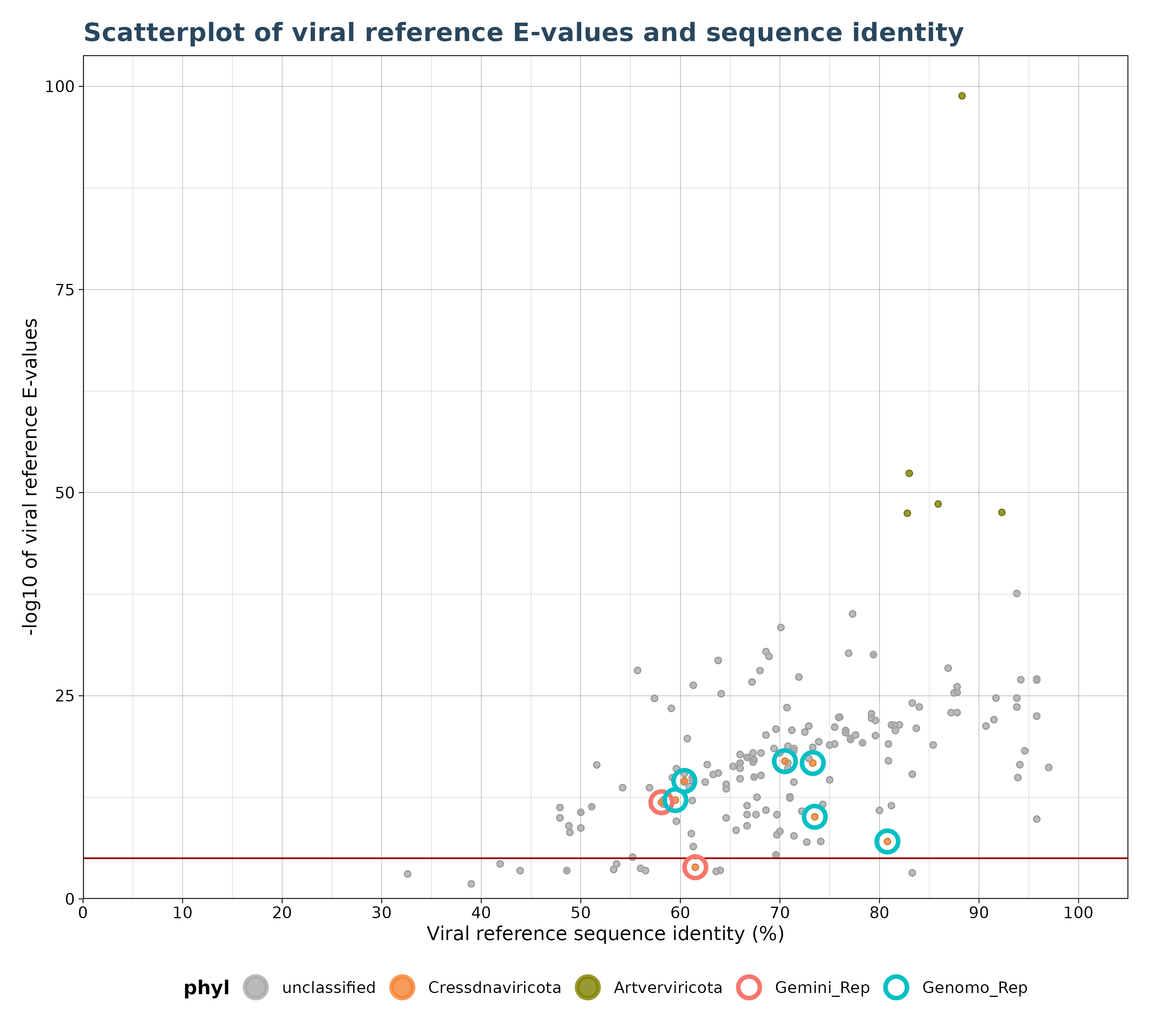
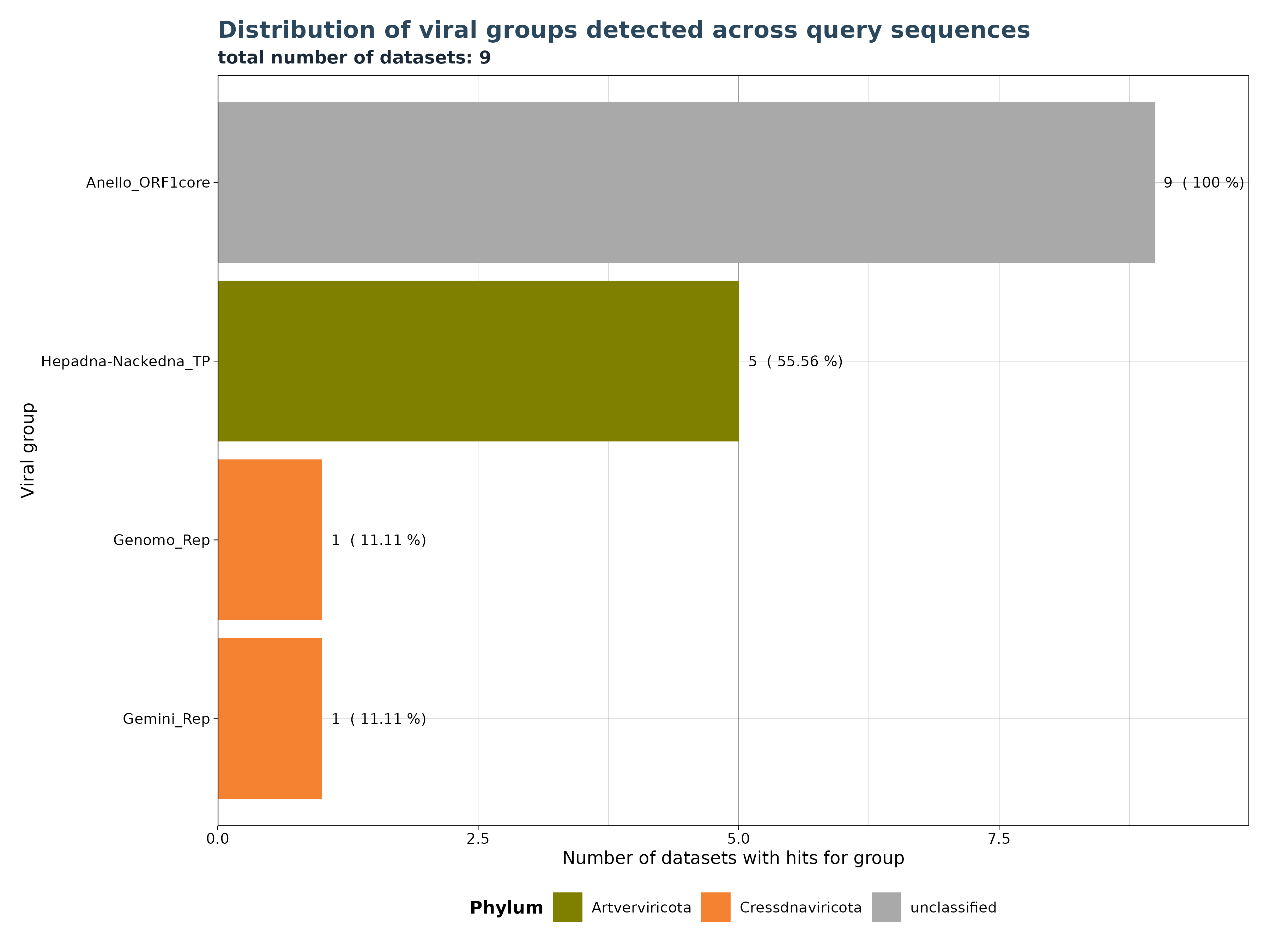
VhgRunsBarplot() takes a VirusHunterGatherer file as
input and plots the distribution of viral groups detected across query
sequences. The cutoff is applied to filter out specific observations
above a specified threshold. This means that if 10 unique SRA-runs (or
local FASTQ Files) detect a viral group, but only 9 have values below
the threshold, then only those 9 will be plotted.
In the example below, we have 9 datasets (SRA_runs). We use “best_query” (groupby) for grouping on the x-axis (here inverted) and see the total number of datasets with hits for each group on the y-axis. Anello_ORF1core is found in all 9 files, - 5 datasets contain observations for Hepadna-Nackedna_TP and both Gemini_Rep and Genomo_Rep have only 1.
### Generate Plot
plot <- VhgRunsBarplot(vh_file,cut = 1e-5)
plot
VhSumHitsBarplot() plots the sum of
reads/micro-contigs/contigs (“best_query”) for each virus group
specified by the groupby argument. The cutoff
value is used to filter out observations above the threshold.
The VhSumHitsBarplot() function generates one of three
bar plot versions, depending on the specified
y_column argument.
Accepted y_column arguments are: - “num_hits”: Number of reads in each group. - “ViralRefSeq_contigs”: Number of micro-contigs in each group. - “contig” (Gatherer Tables only): Number of contigs in each group.
The example below shows that a total of 12704 hits exist in our data, but almost 89 % of the hits mactch to Hepadna-Nackedna_TP, followed by Anello_ORF1core with 11.24 % and less than 1 % for both Genomo_Rep and Gemini_Rep.
### Generate Plot for reads per group
plot <- VhSumHitsBarplot(vh_file,cut = 1e-5,
y_column = "num_hits")
plot$plot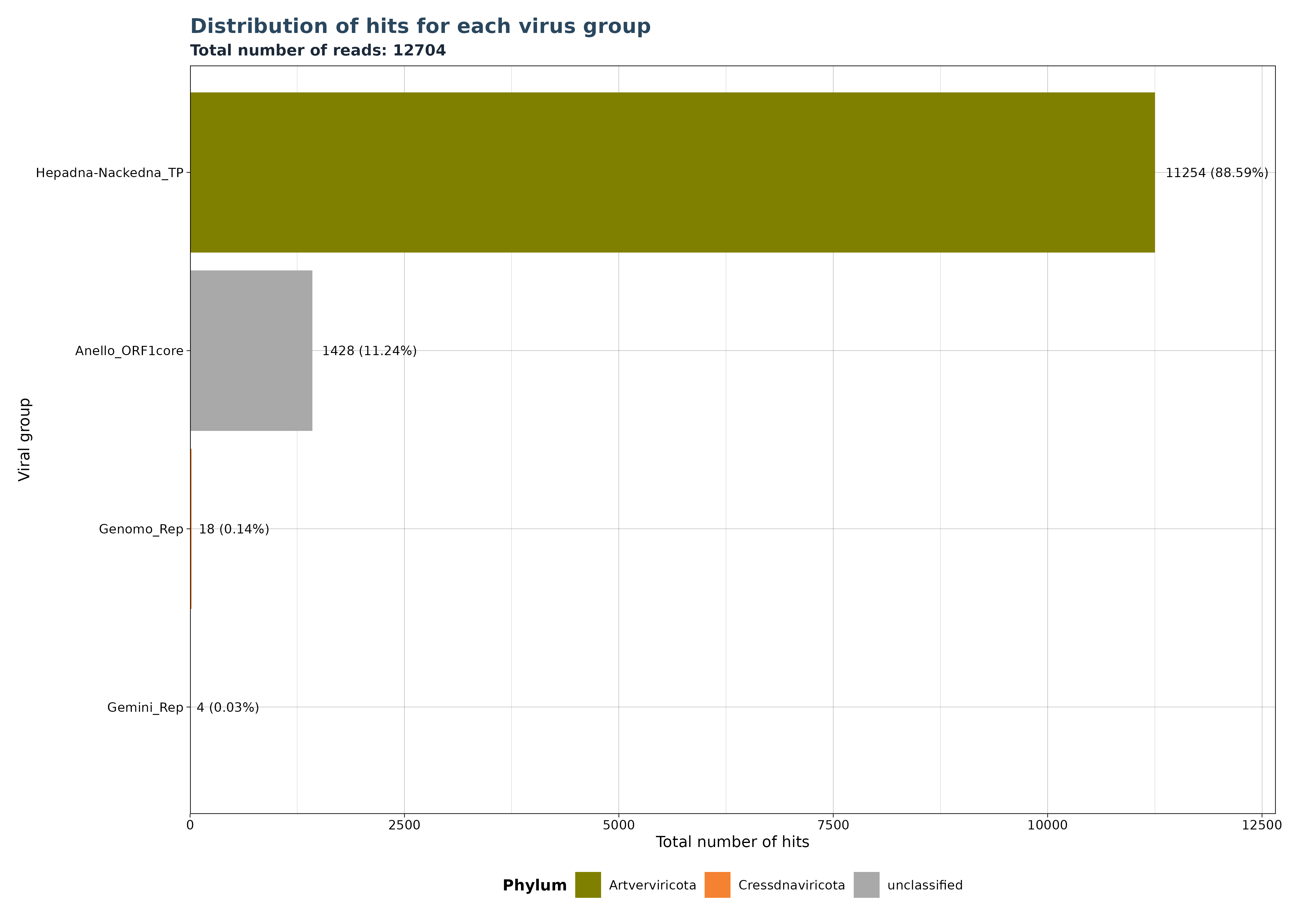
### Generate Plot for micro-contigs per group
plot_micro <- VhgSumHitsBarplot(vh_file,cut = 1e-5,
y_column = "ViralRefSeq_contigs")
plot_micro$plot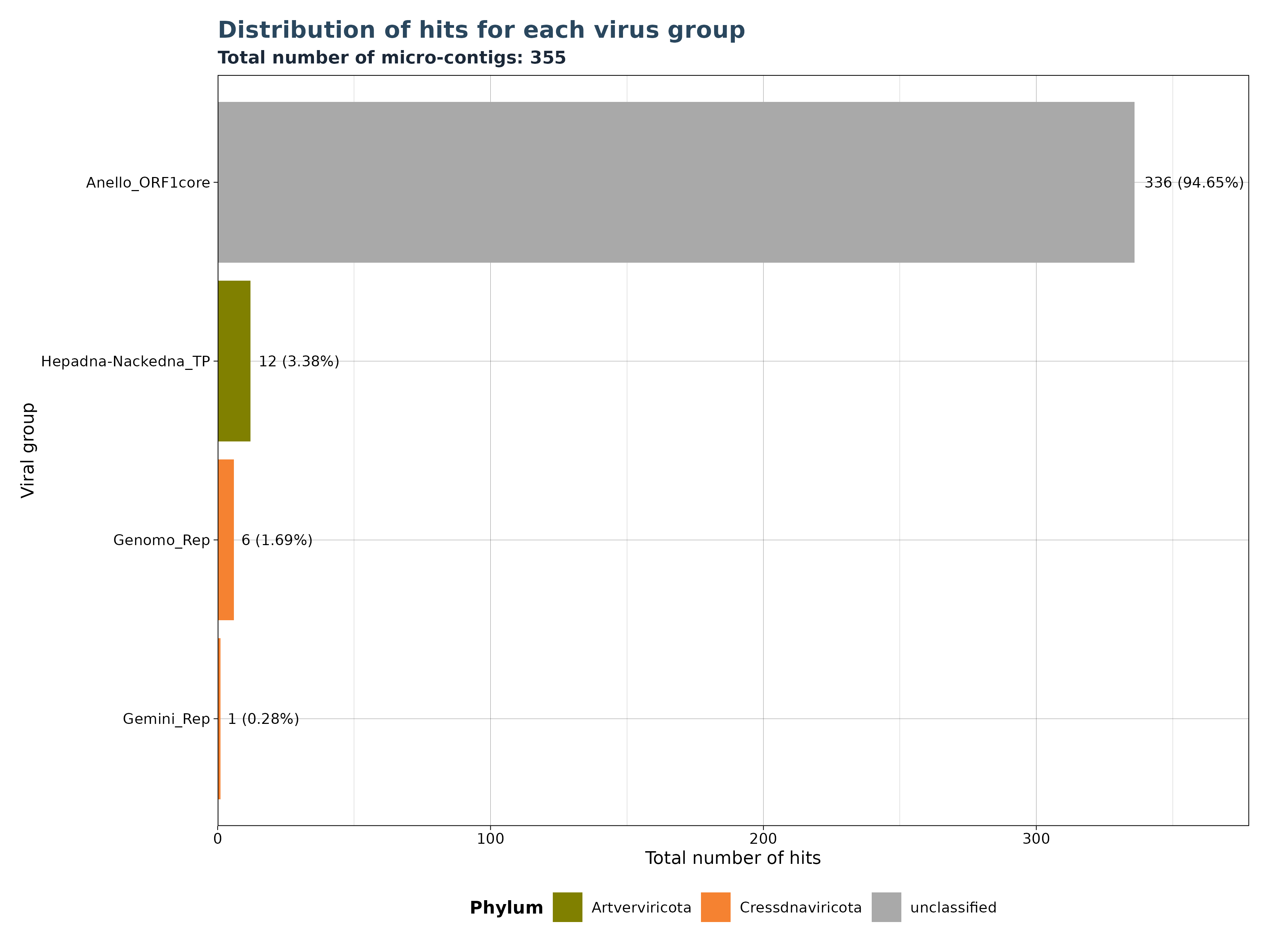
### Generate Plot for assembled contigs per group (Gatherer only)
contig_plot <- VhgSumHitsBarplot(vg_file,groupby = "ViralRefSeq_taxonomy",
y_column = "contig")
contig_plot$plot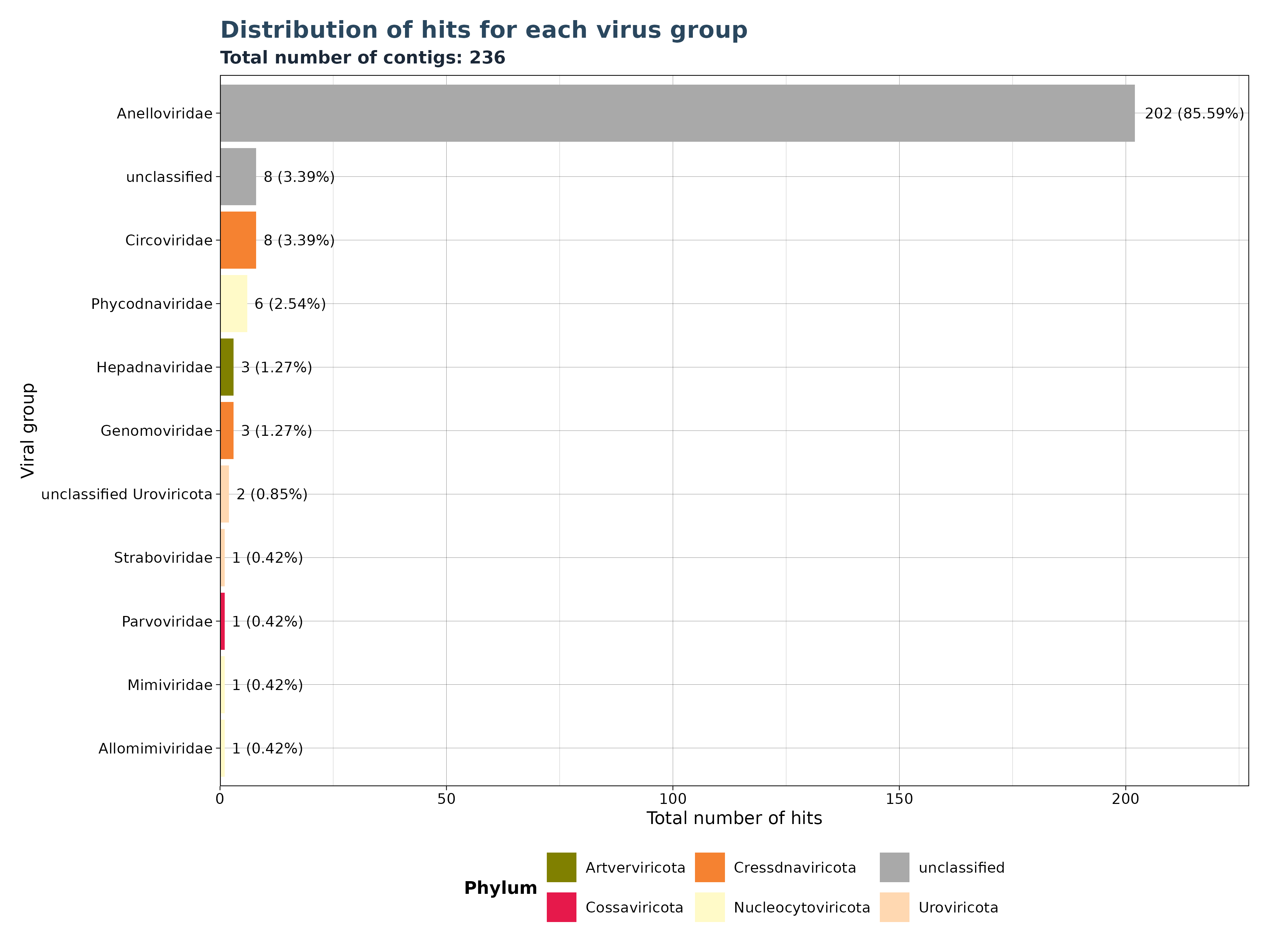
VgConLenViolin() accepts only VirusGatherer hittables as
input and generates a violin plot for contig lengths across viral
groups. Violin plots require at least two data points; if only a single
data point is available, a dot is displayed by default (see plot). Users
can also set a minimum threshold for observations, and groups with fewer
than this threshold are excluded from the plot.
# create a violinplot.
violinplot <- VgConLenViolin(vg_file=vg_file,cut = 1e-5,log10_scale = TRUE)
violinplot$plot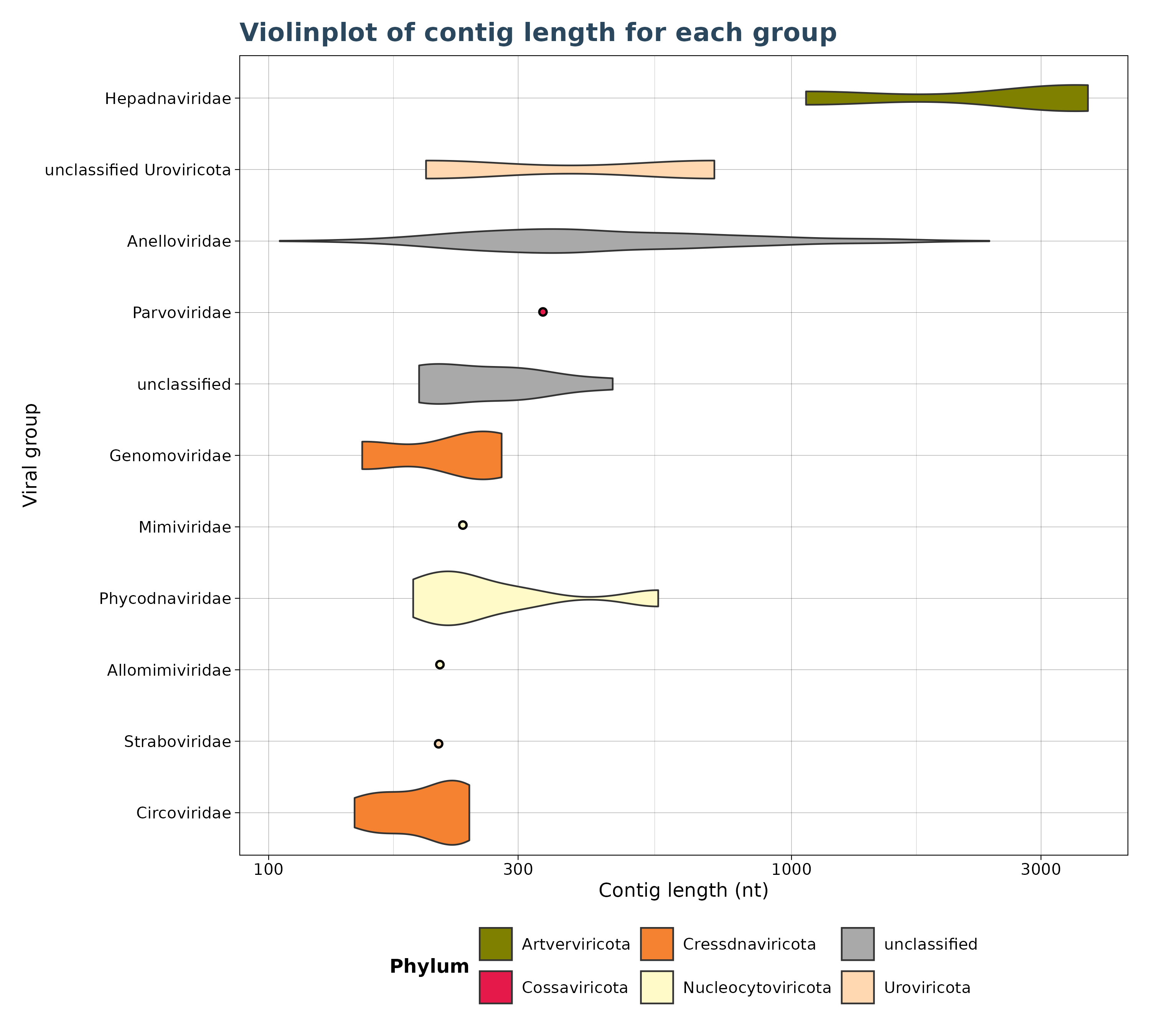
VhgRunsTable() function takes VirusHunter files as input
and generates a graphical table, providing information about which run
has found which virus group. This makes it a valuable complement to the
VhgRunsBarplot() function. While
VhgRunsBarplot() provides information in a plot that
quantifies the number of unique runs finding a virus group,
VhgRunsTable() presents the same information in table form,
showing which runs are found along with their names (SRA accessions,
FASTQ).
VhgRunsBarplot() plots the distribution of viral groups
detected across query sequences, but does not provide information about,
which dataset detects a specific virus group. In the example above, we
see that 5 datasets contain observations for Hepadna-Nackedna_TP, but
only VhgRunsTable() shows which files specifically detect
Hepadna-Nackedna_TP (see table below).
### Generate table with defaul arguments
table <- VhgRunsTable(vh_file,cut = 1e-5)
table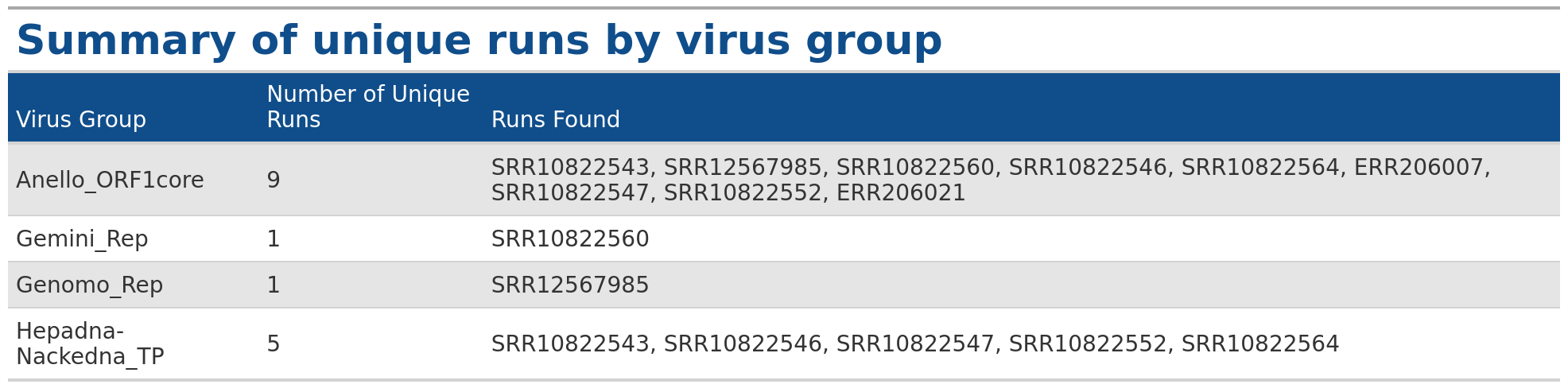
This function creates a formatted table using the GT package, based
on input data with specified column names. It is useful for generating
tables that cannot be produced with VhgRunsTable(). Users
have the option to generate tables with the same styling as those
generated by the VhgRunsTable() function but for the
summary statistics objects generated by the Virusparies plot functions
or the user-defined objects.
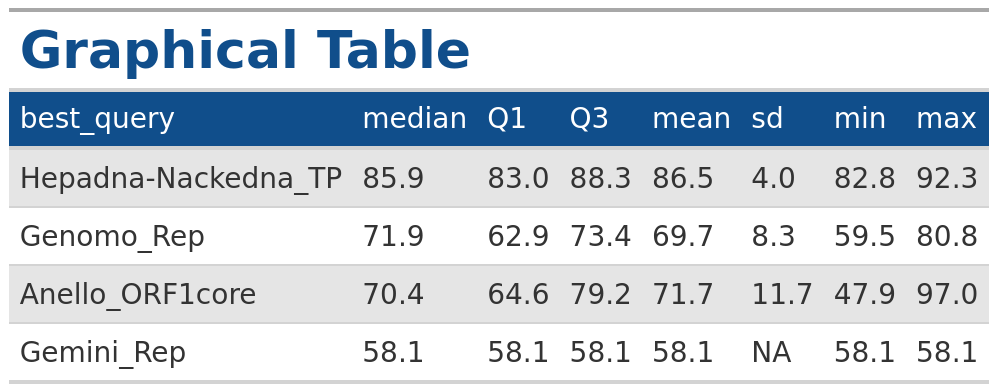
In the example below we generate a table for the summary statistics
output for VhgBoxplot() where
y_column = “ViralRefSeq_ident” with the same
style as the output from the VhgRunsTable() function.
### Plot boxplot for "identity"
identity <- VhgBoxplot(vh_file,y_column = "ViralRefSeq_ident")
# Generate table
VhgTabularRasa(identity$summary_stats)
Processed VirusHunterGatherer hittables and summary statistic tables
can be exported as CSV or TSV files using the
ExportVirusDataFrame() function.
# generate a plot that returns both processed hittables (outlier) and summary stats
plot1 <- VhgBoxplot(vh_file, x_column = "best_query", y_column = "ViralRefSeq_E")
# export hittable as tsv (same format as input hittables)
ExportVirusDataFrame(df=plot1$outlier,file_name="outlier",file_type="tsv")
# export summary stats as csv
ExportVirusDataFrame(df=plot1$summary_stats,file_name="summarystats",file_type="csv")
Plots can be exported in various formats using the
ExportVirusPlot() function. Supported formats include
“eps”, “ps”, “tex”, “pdf”, “jpeg”, “tiff”, “png”, “bmp”, “svg”, and
“wmf” (Windows only). When the device argument is set to NULL, the file
extension in the filename determines the export format.
### Generate Basic plot
plot <- VhgIdentityScatterPlot(vh_file,cutoff = 1e-5)
### Export plot
ExportVirusPlot(plot=plot,file_name="testplot.png",width=8,height=6,units="in")
! Depending on the plot, the final image might be cropped or truncated. Experiment with height, width, and resolution.
The ExportVirusGt() function utilizes the
gtsave() function from the GT package to export graphical
tables in various formats. This feature is currently in an experimental
phase and may not operate as expected. !
Please note that exporting PNG and PDF files requires Google Chrome or a
Chromium-based browser.
### Using first 10 rows of SRA_run,num_hits,bestquery,ViralRefSeq_E and Identity col.
vh_file_part <- vh_file[c(1:10),c(1,7,9,10,11)]
### Generating a gt
table <- VhgTabularRasa(vh_file_part,title = "first 10 rows of vh_file",subtit =
"example for any table",names_ = c("Runs","Number of Contigs","Best Query Result",
"Reference E-Value","Refrence Identity"))
### Export gt as docx file
ExportVirusGt(gtable=table,filename="vh_parttable.docx")Virusparies includes utility functions to process VirusHunterGatherer hittables, extract targeted information, and update the internal ICTV dataset.
Multiple hittables can be combined using the
CombineHittables() function, provided they are of the same
type (e.g., VirusHunter hittables can only be combined with other
VirusHunter hittables).
path <- system.file("extdata", "virushunter.tsv", package = "Virusparies")
file <- ImportVirusTable(path)
file2 <- ImportVirusTable(path) # both files have 180 observations
combined_file <- CombineHittables(file,file2)
print(nrow(combined_file))VhgPreprocessTaxa() extracts user-defined taxonomy
information from the ViralRefSeq_taxonomy column, which contains taxa
data separated by “|” (e.g.,
“taxid:2065037|Betatorquevirus|Anelloviridae”). In the example below, if
the virus family is selected, only families ending with “viridae” (e.g.,
“Anelloviridae”) will remain in the processed ViralRefSeq_taxonomy
column. When family information is unavailable, other taxa details are
compared with the internal ICTV dataset to infer the phylum. If a phylum
is found, the observation is classified as “unclassified” followed by
the phylum name from the ICTV dataset. Otherwise, the term
“unclassified” is used.
VhgPreprocessTaxa() is used internally by various
Virusparies functions. However, as the size of both the hittables and
ICTV dataset increases, the processing time also grows. This makes
VhgPreprocessTaxa() a potential bottleneck in terms of
execution speed. Therefore, we recommend preprocessing taxonomy
information with VhgPreprocessTaxa() before generating
plots or graphical tables for large hittables.
path <- system.file("extdata", "virushunter.tsv", package = "Virusparies")
file <- ImportVirusTable(path)
file_filtered <- VhgPreprocessTaxa(file,"Family")
print("ViralRefSeq_taxonomy before processing:")
print(head(file$ViralRefSeq_taxonomy,5))
#>[1] "taxid:2065037|Betatorquevirus|Anelloviridae"
#>[2] "taxid:2065052|Gammatorquevirus|Anelloviridae"
#>[3] "taxid:2065037|Betatorquevirus|Anelloviridae"
#>[4] "taxid:687379|Gammatorquevirus|Anelloviridae"
#>[5] "taxid:2065046|Gammatorquevirus|Anelloviridae"
print("ViralRefSeq_taxonomy after processing:")
print(head(file_filtered$ViralRefSeq_taxonomy,5))
#>[1] "Anelloviridae"
#>[2] "Anelloviridae"
#>[3] "Anelloviridae"
#>[4] "Anelloviridae"
#>[5] "Anelloviridae"When utilizing Virusparies in your research or software development,
kindly reference the R package using the citation obtained from the
citation() function:
### Citation function
citation("Virusparies")
#> To cite package ‘Virusparies’ in publications use:
#>
#> Ruff S (2024). _Virusparies: Data Visualisations for
#> VirusHunterGatherer hittables output_. R package version
#> 1.0.0, <https://github.com/SergejRuff/Virusparies>.
#> A BibTeX entry for LaTeX users is
#> @Manual{,
#> title = {Virusparies: Data Visualisations for VirusHunterGatherer hittables output},
#> author = {Sergej Ruff},
#> url = {https://github.com/SergejRuff/Virusparies},
#> year = {2024},
#> note = {R package version 1.0.0},
#> }Sergej Ruff formulated the idea behind Virusparies and was responsible for its implementation.
Chris Lauber and Li Chuin Chong from Twincore - Centre for Experimental and Clinical Infection Research provided ideas for improvements. Chris Lauber is the main developer behind the VirusHuntergatherer software and the Group Leader of the Computational Virology working group at the Institute for Experimental Virology, TWINCORE.