![]()
The goal of MetabolomicsBasics is to provide a set of functions to investigate raw data (a matrix of intensity values) from (metabol)omics experiments, i.e. following peak picking and signal deconvolution. Functions can be used to i.e.:
A detailed description of best practice usage may be found in the publication https://link.springer.com/protocol/10.1007/978-1-4939-7819-9_20.
You can install the development version of MetabolomicsBasics from GitHub with:
# install.packages("devtools")
devtools::install_github("janlisec/MetabolomicsBasics")A typical use case would be to compute a Principal Component Analysis:
raw <- MetabolomicsBasics::raw
sam <- MetabolomicsBasics::sam
MetabolomicsBasics::RestrictedPCA(dat = raw, sam = sam, group.col = "Group", legend.x = "bottomleft", medsd = TRUE, fmod = "Group") More
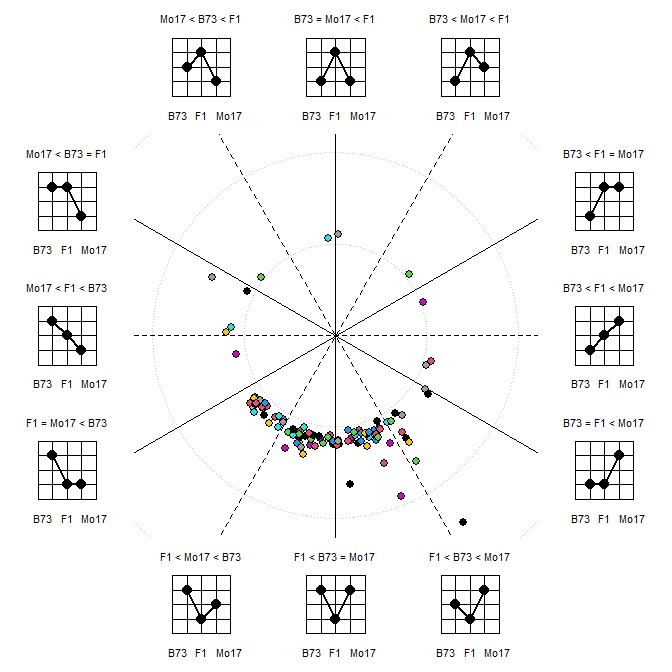
elaborate plots, like the polar coordinate visualization of heterosis
pattern are possible:
More
elaborate plots, like the polar coordinate visualization of heterosis
pattern are possible:
x <- t(raw)
colnames(x) <- sam$GT
MetabolomicsBasics::PolarCoordHeterPlot(x=x, gt=c("B73","B73xMo17","Mo17"), plot_lab="graph", col=1:10, thr=0.5, rev_log=exp(1))
#> Parameter 'col' should be a color vector of length nrow(x)